1. What we do
PacDOCK is a collection of software programs that runs on the web and provide useful tools for comparison of molecular conformations, graphic 3D visualisation and cluster analysis of docking results.
2. Which tools we offer
PacDOCK is composed of three tools:
- ProRMSD, which performs automatic atom matching and RMSD calculation
- PacVIEW, enables 3D graphic visualisation to study and interpret docking results
- ClusDOCK, which performs cluster analysis of docked poses offering three different algorithms
3. Why use PacDOCK
Computational chemistry is nowadays an integrative and fundamental part of pharmaceutical research, particularly in the computer-aided drug design (CADD) field for the development and optimization of pharmaceutical leads.
Molecular docking is one of the most employed techniques in modern drug design and structural biology. The goal of protein-ligand docking is to predict the behaviour of a relatively small molecule (ligand) in the binding site of a biological macromolecule such as a protein or a nucleic acid (target).
For this purpose, the pose of the ligand when it is bound to a target must be determined. This consist of its position (x-, y-, and z-translations), its orientation (Euler angles, axis-angle, or a quaternion), and, if the ligand is flexible, its conformation (the torsion angles for each rotatable bond). The pose is defined according to protein and ligand shape, but also on the basis of protein-ligand intermolecular interactions, which altogether permit establishing a docking score, reliant on binding affinity.
In order to perform a docking study, a 3D structure of the protein is required. The number of protein structures determined experimentally using X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy or cryogenic electron microscopy (cryo-EM) has greatly increased over the last two decades, helping a wide use of docking strategies.
Molecular docking involves computationally exploring a search space that is defined by the molecular representation used by the method, and ranking candidate solutions to determine the best binding mode. Thus, docking requires both a search method and a scoring function.
The ability of docking programs to accurately assess binding affinity is dependent on their ability to find the optimal pose of the ligand in the protein binding pocket, thus docking programs are often benchmarked by their ability to reproduce the pose of a ligand from a known experimental protein-ligand complex structure, in a process called “re-docking”.
Anyway, comparing docking methods is not that easy since some methods can perform better with certain classes of ligands and targets, thus a good modus operandi is to try several docking methods to determine the best one for the specific problem.
To measure the ability of a docking program to reproduce the experimental pose, the root-mean-square-deviation (RMSD) of Cartesian coordinates of atomic position can be involved by performing a comparison between the predicted pose and the reference pose of the ligand.
RMSD is one of the most useful features for structural comparison, widely used in protein-ligand docking and molecular dynamics studies. With the use of RMSD, a single value can provide information on the average deviation of a binding pose with respect to a reference orientation and conformation in the 3D space.
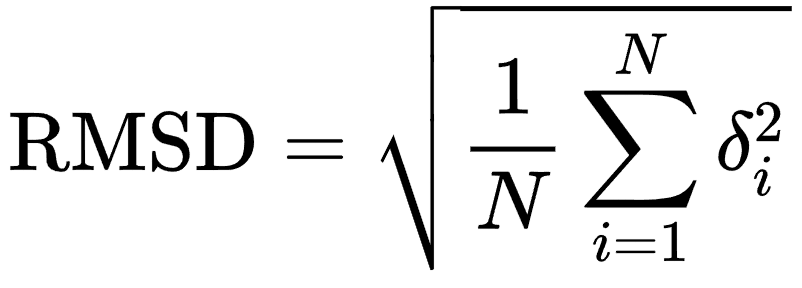 where N is the number of atoms in the ligand, and δi is the Euclidean distance between the ith pair of corresponding atoms
It seems very easy to calculate RMSD, but this formula only works with the assumption of a direct atomic correspondence between molecular structures.
where N is the number of atoms in the ligand, and δi is the Euclidean distance between the ith pair of corresponding atoms
It seems very easy to calculate RMSD, but this formula only works with the assumption of a direct atomic correspondence between molecular structures.
For overcame this problem, ProRMSD has been developed, since it performs an automatic atom matching and RMSD calculation.
> For more information about ProRMSD, see ProRMSD documentation.
Positional measurements, such as the RMSD, must be interpreted with caution and can be combined with measures based on interaction and visual inspection of docking results. The interaction-based measure consists of the investigation of the key interactions that the ligand forms with its receptor.
PacVIEW can provides receptor-ligand interactions, which allows a complete and more reliable interpretation of docking results.
In addition, by using PacVIEW, the numerical value of RMSD between poses can be associated with a visualisation of the spatial differences of the docking result compared to the reference pose. Thus, PacVIEW is an important integrative tool that can be used after performing a RMSD calculation with ProRMSD.
This tool has the benefit of being a free web viewer that does not require the installation of any packages.
> For more information about PacVIEW, see PacVIEW Documentation.
Once the accuracy of the docking method has been assessed and several docked poses have been computed, it could be useful to cluster the results of a docking calculation in order to determine the most probable binding poses.
The docking results are evaluated by a scoring function that, depending on the docking program, consider the geometrical and chemical complementarity between the protein and the ligand. If different binding poses with nearly equal scoring functions are determined, or the simulation has been run multiple times (a common case when stochastic docking methods are used), the similarity of the predicted binding modes can be assessed by performing cluster analysis. Cluster analysis defines a set of techniques that aims to group data into categories, called clusters, on the basis of their similarities.
Clustering reduces the number of poses that need to be examined since further analyses need only consider a representative pose from each cluster. To perform this, a matrix of pairwise RMSD values is calculated and docked poses are clustered according to an RMSD cut-off.
To execute a clustering of docked poses, ClusDOCK has been developed and it allows the clustering with three different algorithms.
> For more information about ClusDOCK, see ClusDOCK documentation.
Molecular docking is one of the most employed techniques in modern drug design and structural biology. The goal of protein-ligand docking is to predict the behaviour of a relatively small molecule (ligand) in the binding site of a biological macromolecule such as a protein or a nucleic acid (target).
For this purpose, the pose of the ligand when it is bound to a target must be determined. This consist of its position (x-, y-, and z-translations), its orientation (Euler angles, axis-angle, or a quaternion), and, if the ligand is flexible, its conformation (the torsion angles for each rotatable bond). The pose is defined according to protein and ligand shape, but also on the basis of protein-ligand intermolecular interactions, which altogether permit establishing a docking score, reliant on binding affinity.
In order to perform a docking study, a 3D structure of the protein is required. The number of protein structures determined experimentally using X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy or cryogenic electron microscopy (cryo-EM) has greatly increased over the last two decades, helping a wide use of docking strategies.
Molecular docking involves computationally exploring a search space that is defined by the molecular representation used by the method, and ranking candidate solutions to determine the best binding mode. Thus, docking requires both a search method and a scoring function.
The ability of docking programs to accurately assess binding affinity is dependent on their ability to find the optimal pose of the ligand in the protein binding pocket, thus docking programs are often benchmarked by their ability to reproduce the pose of a ligand from a known experimental protein-ligand complex structure, in a process called “re-docking”.
Anyway, comparing docking methods is not that easy since some methods can perform better with certain classes of ligands and targets, thus a good modus operandi is to try several docking methods to determine the best one for the specific problem.
To measure the ability of a docking program to reproduce the experimental pose, the root-mean-square-deviation (RMSD) of Cartesian coordinates of atomic position can be involved by performing a comparison between the predicted pose and the reference pose of the ligand.
RMSD is one of the most useful features for structural comparison, widely used in protein-ligand docking and molecular dynamics studies. With the use of RMSD, a single value can provide information on the average deviation of a binding pose with respect to a reference orientation and conformation in the 3D space.
where N is the number of atoms in the ligand, and δi is the Euclidean distance between the ith pair of corresponding atoms
For overcame this problem, ProRMSD has been developed, since it performs an automatic atom matching and RMSD calculation.
> For more information about ProRMSD, see ProRMSD documentation.
Positional measurements, such as the RMSD, must be interpreted with caution and can be combined with measures based on interaction and visual inspection of docking results. The interaction-based measure consists of the investigation of the key interactions that the ligand forms with its receptor.
PacVIEW can provides receptor-ligand interactions, which allows a complete and more reliable interpretation of docking results.
In addition, by using PacVIEW, the numerical value of RMSD between poses can be associated with a visualisation of the spatial differences of the docking result compared to the reference pose. Thus, PacVIEW is an important integrative tool that can be used after performing a RMSD calculation with ProRMSD.
This tool has the benefit of being a free web viewer that does not require the installation of any packages.
> For more information about PacVIEW, see PacVIEW Documentation.
Once the accuracy of the docking method has been assessed and several docked poses have been computed, it could be useful to cluster the results of a docking calculation in order to determine the most probable binding poses.
The docking results are evaluated by a scoring function that, depending on the docking program, consider the geometrical and chemical complementarity between the protein and the ligand. If different binding poses with nearly equal scoring functions are determined, or the simulation has been run multiple times (a common case when stochastic docking methods are used), the similarity of the predicted binding modes can be assessed by performing cluster analysis. Cluster analysis defines a set of techniques that aims to group data into categories, called clusters, on the basis of their similarities.
Clustering reduces the number of poses that need to be examined since further analyses need only consider a representative pose from each cluster. To perform this, a matrix of pairwise RMSD values is calculated and docked poses are clustered according to an RMSD cut-off.
To execute a clustering of docked poses, ClusDOCK has been developed and it allows the clustering with three different algorithms.
> For more information about ClusDOCK, see ClusDOCK documentation.
References:
2. Meng,X.-Y., Zhang,H.-X., Mezei,M. and Cui,M. (2011) Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr. Comput. Aided-Drug Des., 7, 146–157.
3. Morris,G.M. and Wilby,M.L. (2008) Molecular Docking. In Kukol,A. (ed), Molecular Modeling of Proteins, Methods in Molecular Biology. Humana Press, Totowa, NJ, Vol. 443, pp. 365–382. 4. Everitt,B.S., Landau,S., Leese,M. and Sthal,D. eds. (2011) Cluster Analysis 5th ed. Wiley Publishing.